EFPIA’s Regulatory Road To Innovation – Back To The Cutting Edge
Executive Summary
Ahead of the first review of European pharma legislation in almost 20 years, senior executives at Lilly and MSD assess key recommendations from EFPIA’s Regulatory Road to Innovation to put the EU regulatory system back at the cutting edge of innovation.
For the first time in almost two decades, this year the European Commission (EC) is reviewing the region’s general pharmaceutical legislation following a consultation period that ended in 2021. The timing is opportune, as the COVID-19 pandemic has drawn attention to issues within life sciences that previously had not been as visible or prevalent. Additionally, in response to the changing global environment, certain pharmaceutical processes were adapted and new technologies implemented, which will have long-term impacts on the industry.
As a key voice in the pharma space, in response to the consultation period the European Federation of Pharmaceutical Industries and Associations (EFPIA) created a ‘Regulatory Road to Innovation’. This provided analysis of the current state of the European market and recommendations to improve its global standing. “The pharmaceutical strategy really provides us with a once in a generation opportunity to take learnings from the pandemic, to make sure that we have a competitive regulatory framework that really supports innovation and responds very quickly to patient needs,” says Alan Morrison, vice president of international regulatory affairs at MSD and chair of EFPIA’s Regulatory Strategy Committee.
Within EFPIA’s roadmap there are 12 recommendations, with four proposals requiring legislative change which they feel should be implemented in the EC’s new framework. Nathalie Moll, director general at EFPIA states, "As an industry, we have developed a series of proposals contained in the Regulatory Road to Innovation that will make Europe more attractive for medical research, development and advanced manufacturing, resulting in earlier availability of innovative therapies to European patients. The proposals are supported by robust data and build on the practical experience of our members and will make a difference for patients if considered in the ongoing review of legislation."
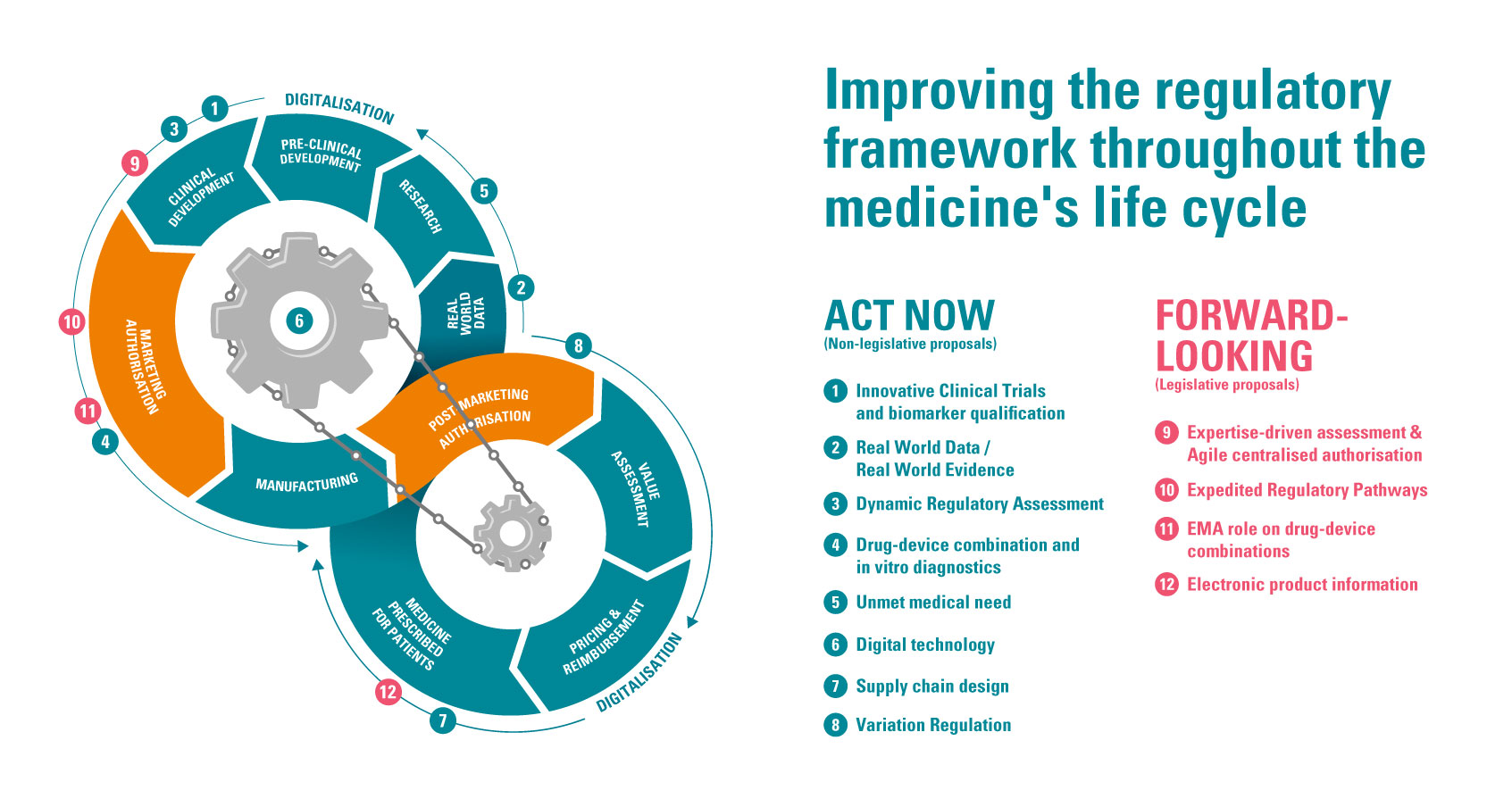
Accelerating Regulatory Decision-Making
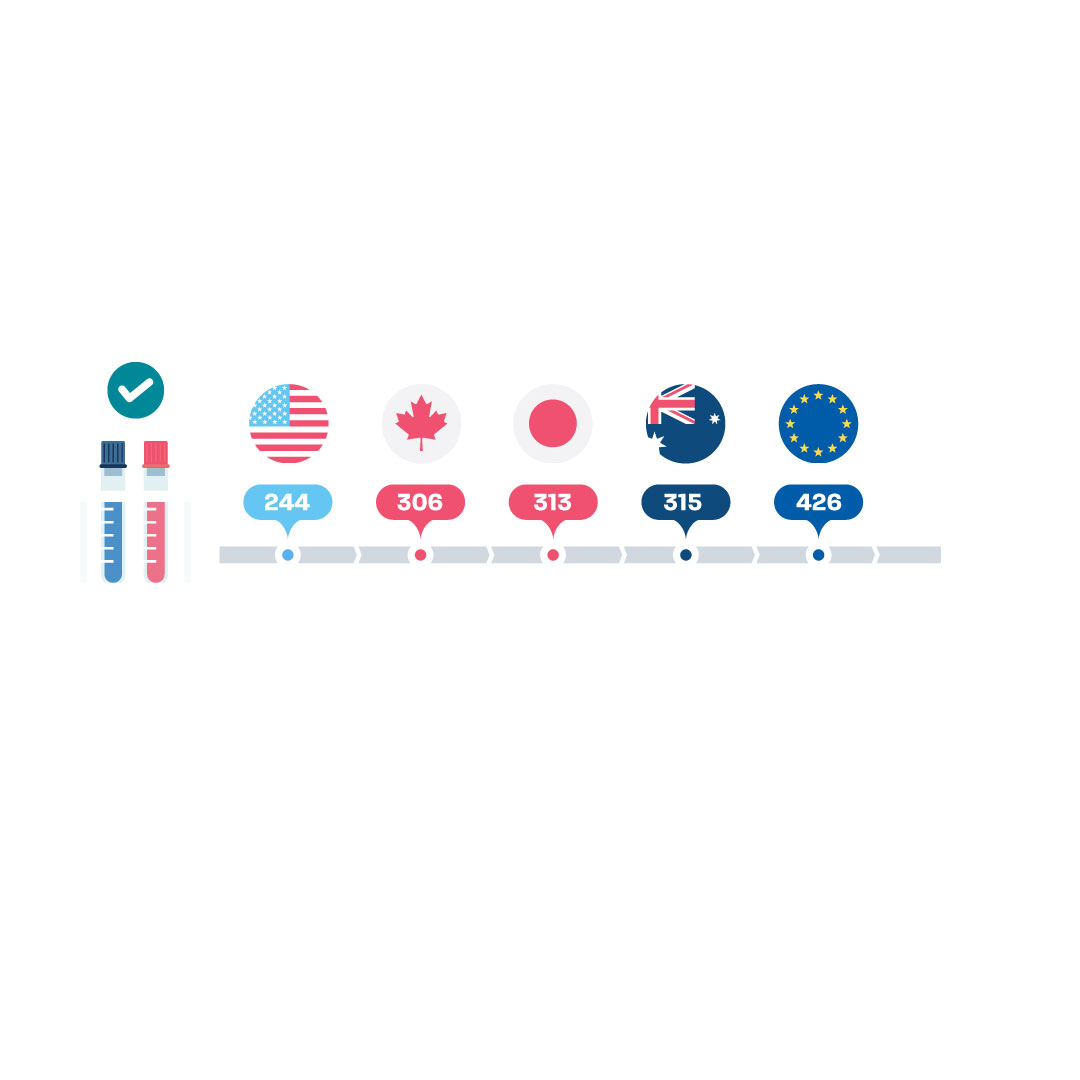
Regulatory decision-making in Europe lags behind other key pharma markets, notes Susan Forda, vice president of international regulatory affairs at Lilly and member of EFPIA’s Regulatory Strategy Committee. “The median time to approval of a new medicine by the European Medicines Agency (EMA) was 426 days, compared with the FDA’s median time of 244,” says Forda, referencing a 2021 report from the Centre for Innovation in Regulatory Science (CIRS).1 The data also show that Europe is not just significantly behind the US, but also over 100 days slower than Health Canada (306 days), Japan’s PMDA (313 days) and Australia’s TGA (315 days).
A specificity of the EU approval system for a new medicine is the lag between a positive CHMP opinion and a formal, legally binding EC decision, which currently takes 67 days on average. These long timeframes are not only frustrating for sponsors on a financial level, but most importantly they have real impact for patients. “From a recent survey conducted by EFPIA on 11 oncology products,2 these 67 days translated into around 18,600 years of potential life lost, which is huge,” states Morrison.
During the pandemic there were instances where this 67-day period could be compressed into just one day, due to the overwhelming global need for COVID-19 vaccines. While these were unprecedented circumstances, it proves that timelines can be reduced. “What we saw during the COVID-19 crisis were a number of initiatives where reviews could be sped up without compromising patient safety,” says Forda.
EFPIA’s Regulatory Road to Innovation suggests as its first proposal that the process between CHMP opinion and EC decision could be made more efficient. Morrison continues, “Firstly, by conducting linguistic and decision-making phases in parallel following CHMP opinion, marketing authorization could be granted 12 days earlier which translates into around 3,300 life years saved. Secondly, deploying modern technologies to facilitate translation could shorten by 10 days, saving 2,500 life years. Finally, there’s opportunity to shorten the written procedure in cases where standing committee checks and balances foresee no objections. This could shorten the decision-making phase by 15 days, saving 4,200 life years.2”
The Role Of Expedited Review Pathways (ERPs)
While the EMA has made steps towards expediting the regulatory process, implementing the PRIME scheme in 2016, successful uptake has been limited. Forda notes that this also sets the EU behind the US: “Only 25% of PRIME applications have been accepted by EMA, while breakthrough designation in the US has had twice as many applications in the last six years and have accepted over 40%.”
EFPIA concludes that lack of clarity surrounding eligibility is a key barrier. Currently, PRIME is limited to molecules that cover an unmet medical need or offer a major advantage over existing treatment options. However, what constitutes an unmet medical need can be subjective. “What we have found is that at the moment, the term of unmet medical need is inconsistently defined within and across organizations and jurisdictions,” says Forda. Positively, the EC has expressed interest in having a common definition, but Forda notes that it will be important that this is thought about carefully. She continues, “It is very important that all relevant stakeholders, particularly patients, are involved in determining what constitutes an unmet medical need and that it applies from drug delivery through to patient access. We would very much appreciate that it be either a broad definition applicable in all contexts, or a description of the context followed by the definition.”
As well as solidifying the unmet medical need definition, EFPIA also proposes that a wider suite of ERPs be implemented to support and incentivize the development of increasingly innovative treatments. These pathways should also be more aligned with global expedited pathways to facilitate harmonized regulatory submission strategies across regions. Forda also points out that this should be extended to post-approval pathways. “We have almost no expedited reviews in the post-approval setting, so not only do we fall down somewhat in first approvals, but we do later too.”
It is critical that these pathways – including PRIME – are equipped with the necessary resources and scientific expertise to facilitate accelerated timelines. EFPIA proposes in its roadmap that PRIME be embedded into legislation, which would enable EMA permanent staff to be allocated to it. “An important point to raise is that the number of EMA staff has remained fairly flat over the last five years, so clearly it’s key to make sure any future model, committee structures and the overall regulatory framework is appropriately resourced,” states Morrison.
Alignment For Combination Products
The lines between the pharma and medical device worlds are blurring, as it is estimated around 25% of the industry pipeline is made up of combination products.3 With this growth only set to continue, it is important the EC gives it due consideration to futureproof new legislation.
Currently, there are two distinct processes for drug and device product approvals. This differs from the approach in the US, where FDA assesses applications for combination products in their entirety. In order to create a more effective pathway, EFPIA believes it is important to create a new legal category for combination products so that they can be regulated in the same way as medicinal treatments. “Having at least a combination product definition that is anchored in legislation would allow the EMA to have a broader oversight in their assessment, and a single regulator approach which would provide a higher degree of certainty and allow for a more coordinated process,” Morrison asserts.
Another critical benefit to entrenching a definition and process for combination products is that it would drive an extended EMA remit, which would warrant the provision of staff with necessary scientific and technical expertise. Morrison continues, “It is really key for the EMA and overall EU regulatory network to ensure they are upskilling their staff and expertise and that the appropriate resources are committed to these regulatory processes of the future.”
Increasing Digitalization For Efficiency And Safety
As day-to-day processes become increasingly digitalized, it is only natural that pharma follows this trend. The pandemic has greatly accelerated this, proving that where industry once thought paper and in-person interaction was unavoidable, that might no longer be the case.
Forda believes there is real opportunity for Europe to lead the way with implementation of more technologically advanced processes. “It would be great to see the EU become a global leader in decentralized clinical trials, because this supports diversity of patients. Studies have shown under 5% of eligible patients are currently able to participate, and in particular racial minorities are underrepresented. Through increased use of digital interactions, we can improve outreach to these diverse groups.”
Another area where EFPIA believes efficiency could be gained by going digital is product information. Its final legislative proposal advocates a move towards transitioning from mandatory paper medicine leaflets to electronic product information (ePI). There are a number of key benefits to this, with the first being for patient safety. “ePI ensures that health care professionals, pharmacists, patients and their carers have access to the latest regulatory information without delay to support. Currently, safety label updates can take up to six months to be implemented,” says Morrison. Other advantages include the possibility to mitigate shortages and enable faster distribution, as well as minimize packaging waste, which ties in with supporting sustainability targets.
In response to concerns that ePI reduces accessibility for patients who do not have access to electronic devices, EFPIA encourages a gradual transition that would be centered around health care professional administered products in the first instance. Morrison notes, “This would build upon several of the ongoing pilots that are currently under development in some European Member States.”
Making Europe Better For Patients
Overall, the hope is that changes to the legislative framework in Europe will make it more globally competitive, as it has lost standing in recent years to other regions as a primary launch market. Equally as important though is the knock-on impact this has on patient prospects. EFPIA hopes that with quicker, more efficient and effective processes across the drug development and regulatory pathways, innovation can flourish and European patients will have access to the best treatment options. “We should really make sure that with the pharmaceutical legislation review, we seize on this opportunity to create an attractive innovation ecosystem for the next generation of medicine,” asserts Morrison. Forda concurs, stating, “The EU regulatory system and its underlying legal framework shouldn’t only be effective for today, but for tomorrow as well. The accelerations, communication and cooperation with companies during the pandemic should become standard and best practice. This will enable us to deliver faster access to all patients in Europe.”
References
1. Centre for Innovation in Regulatory Science, CIRS RD Briefing 81 – New drug approvals in six major authorities 2011-2020 (2021) https://cirsci.org/publications/cirs-rd-briefing-81-new-drug-approvals-in-six-major-authorities-2011-2020/
2. EFPIA, Every Day Counts - Improving Regulatory Timelines To Optimise Patient Access To Innovative Oncology Therapies In Europe (2021) https://www.efpia.eu/media/636486/improving-regulatory-timelines-to-optimise-patient-access-to-innovative-oncology-therapies-in-europe.pdf
3. EFPIA, Evidence MIX (Measures, Insights, and eXamples): Evaluating the EU Regulatory System (2021) https://www.efpia.eu/media/636564/evidence-mix_final-9-dec-2021.pdf